Hi all,
I’m really new to bioinformatics and this is the first time I’ve used mothur toolsuite on Galaxy, so please excuse my lack of knowledge and ability to troubleshoot.
I’m following the 16S Microbial Analysis with mothur Galaxy tutorial (16S Microbial Analysis with mothur (extended)) using a dataset I got from a paper article (paired-end reads generated from illumina-miseq)
I followed the tutorial closely but skipped the part involving error rate calculation using mock community.
In the tutorial, they used the files generated from the mock community section. Since I didn’t do this part, I didn’t get the files for that part.
Instead, I used the most recent files from the tool remove.lineage (fasta, tax, and count files)
with a cutoff of 0.03 and taxlevel of 4.
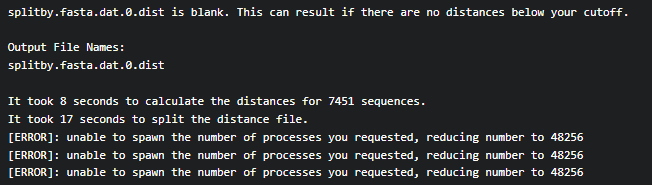
Doing so gave me an error which I have no idea how to troubleshoot (after looking at the tool error guide on Galaxy: Job and Tool Error Help - Galaxy Community Hub).
Here is the error:

Any help would be greatly appreciated! Thank you.