i am new to mothur. Finished analysing the data using Mothur SOP for Iontorrent.
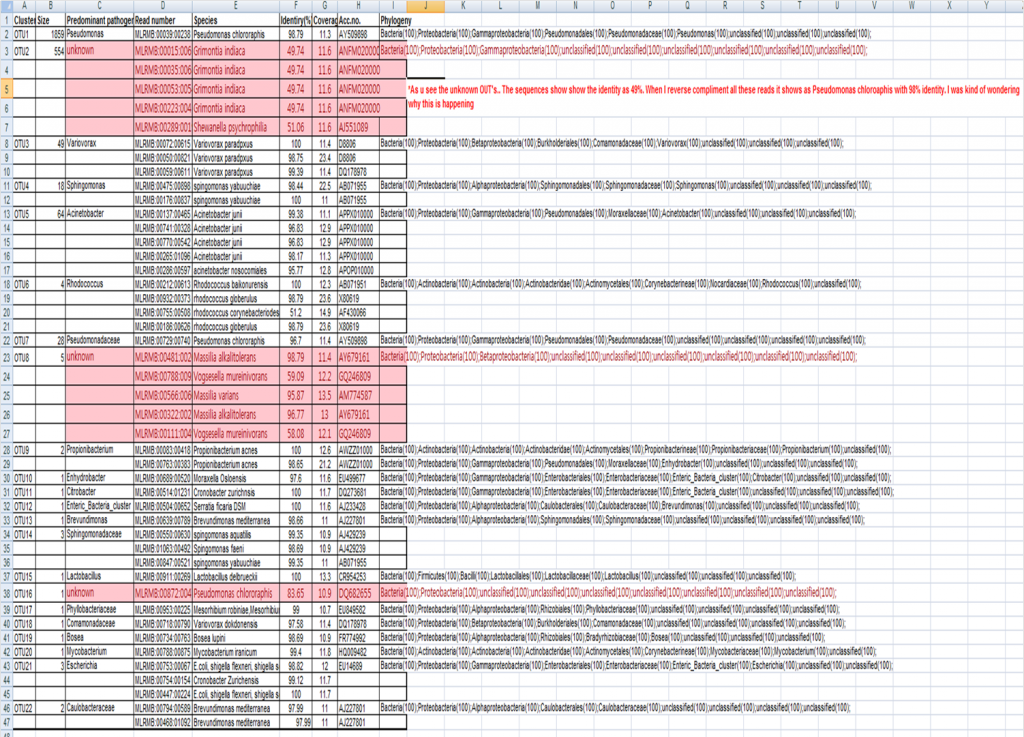
However, there is one OTU consists of 500 sequence which is unclassified. i picked up the individual reads belonging to this OTU, and cheked it in Eztaxon.com for taxonomy identification. It showed 50% similarity threshold. However, when i reverse translate these reads it gives me the 98 to 99% similarity to pseudomonas species. when i reverse translate all the reads belonging to this OTU, it gives 98% similairty.
while aligning sequences i had used the command flip=T.
I’ve never heard of Eztaxon.com and the link appears to be dead. It could be that their database is the reverse complement (not reverse transcribed). Why not just use classify.seqs with one of the reference databases we supply?
We had used the classify.seqs with the mother database itself…
here is the command “mothur> classify.seqs(fasta=ncr.trim.unique.good.filter.unique.precluster.pick.fasta, name=ncr.trim.unique.good.filter.unique.precluster.pick.names, group=ncr.good.pick.groups, template=silva.bacteria.fasta, taxonomy=silva.bacteria.silva.tax, cutoff=80, processors=2)”
The OTU2 is unclassified.The individual read hit shows its Grimontia indiaca with 49% identity…but then when we reverse compliment it, it shows 98% identification with Pseudomonas chlororaphis in the data base.
Sorry, but it’s really not clear what your process is for getting to the point of having sequences to classify. I suspect that you either sequenced from right to left or in trim.seqs you used flip=T when you didn’t mean to. If you use flip=T in classify.seqs it will try both directions if you get a bad assignment.