Mothur community (esp those of you who are running the MiSeqs).
Has anyone seen good read 1, indices, and horrible read 2? I had one particular set of samples fail several times with that exact pattern. Illumina wasn’t super supportive because the read 2 sequencing primer is 62-66, they are saying that is too low. But I’m using the same primers as the vast majority of people doing v4 microbiome work. So, I got them to give me a kit to try to run the samples on another miseq where that library finally worked. The success on the other machine triggered a visit by a FAE who replaced my temp control module. But now my first run after the repair has the same pattern (read one and the indices are great, read 2 sucks).
Hmmm, we aren’t seeing that. But our experience with Illumina is that you have to be persistent with them and agree to do the various PhiX control runs, etc. We’ve had a number of problems with optics alignments, fluidics, complexity, etc. where each step of the way they blamed our PCR set up when it was really a hardware or chemistry problem.
My last run was successful. They recommended that I double the amount of sequencing primers-thinking that the primers aren’t sticking as well since they are just below the annealing temp. I used 6.5ul 100mM of each primer, spiked into wells 12, 13, 14. Extra primers is a pretty cheap solution if that’s really all that is going on.
thanks, I’d been following that thread-our failures were different. I posted about them in more detail over there before I came to conclude that it might be sequencing primer related. If anyone wants to see exactly what kind of failures I was seeing http://seqanswers.com/forums/showthread.php?t=65825
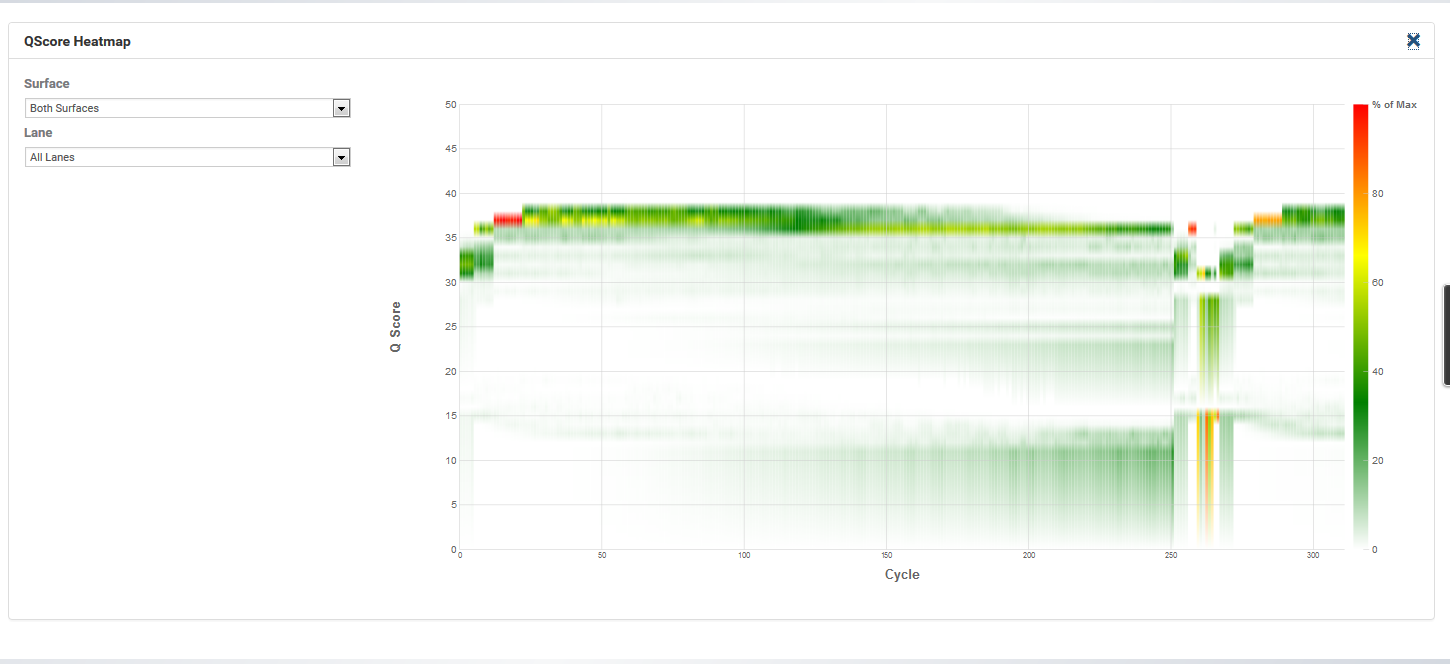
I have the same issue. We are sequencing at the same facility for a while now, and our first run last year was good. I now got data for two more MiSeq V4 runs, both have issues in the start of the reverse read. The first run has 3 Ns in the same positions, but the second run reverse read is almost all Ns for the first 50 odd bases. Some bases (always the same in all reads) are not Ns though, I guess those are more variable? The all look like this:
The guy running the sequencer says this is always a problem with the V4 primers, the sequences are too homogeneous at the start of the reverse read. He suggested to switch to V1/3, or use those staggered primers from the Fadrosh paper. But there must be dozens of people using this primer set, as you guys published in the Kozich paper, successfully ?
I was about to order a full set of the V4 primers with dual barcodes, now I’m not sure what to do. I’d be happy for any advice what they can change when running these samples on the MiSeq again. I’ll ask them about doubling sequencing primer amounts, but we don’t have so much money that we can just run a bunch of tests, either, unfortunately.
Your q30 plot looks a little different but has a similar weird pattern of q score drops. Have you read through this thread? http://seqanswers.com/forums/showthread.php?t=65825 It has all the details of what we tried. My FAS will be here later today, I’ll ask if she’s able to talk to someone who isn’t her direct responsibility about a similar issue to what I had. PM me with your contact info if you’d like to try to talk to her.

{kind=link}