Hi all,
I am following the MiSeq SOP and ran the get.oturep command:
get.oturep(list=final.tx.list, count=stability.contigs.good.count_table, method=abundance)
The list file I used is phylotype based.
For the output generated, can I assume that the order of sequences follows the same order of # of phylotypes so that I can align them with the taxonomy file?
When I ran the command on previous versions of mothur, the output would generate a sequence name and OTU# for each OTU that I could align with the taxonomy file. Then based on the OTU I am looking for, I could search the target sequence in the fast file and search the sequence on blast.
Thank you,
Christelle
Hi Christelle,
Yes, the orders should be the same.
Thanks,
Pat
Thank you for the help with this!
I aligned the rep sequences with the phylotype taxonomy file and I’m using the stability.trim.contigs.good.fasta to look up the sequence. When blasting the sequence on NCBI, for example, for Petromonas, the sequence identified belonged to Wenzhouxiangella sediminis strain XDB06, which I didn’t expect. Another example, for Syntrophomonas, the rep sequence blasted was identified for Methanothrix.
I am not sure where the error occurred and would appreciate additional help!
Thank you,
Christelle
Can you post some examples of output from your files along with the names of the files? Also, if you use classify.otu you should be able to get the classification of each of the phylotypes without having to use get.oturep.
Pat
The files I used:
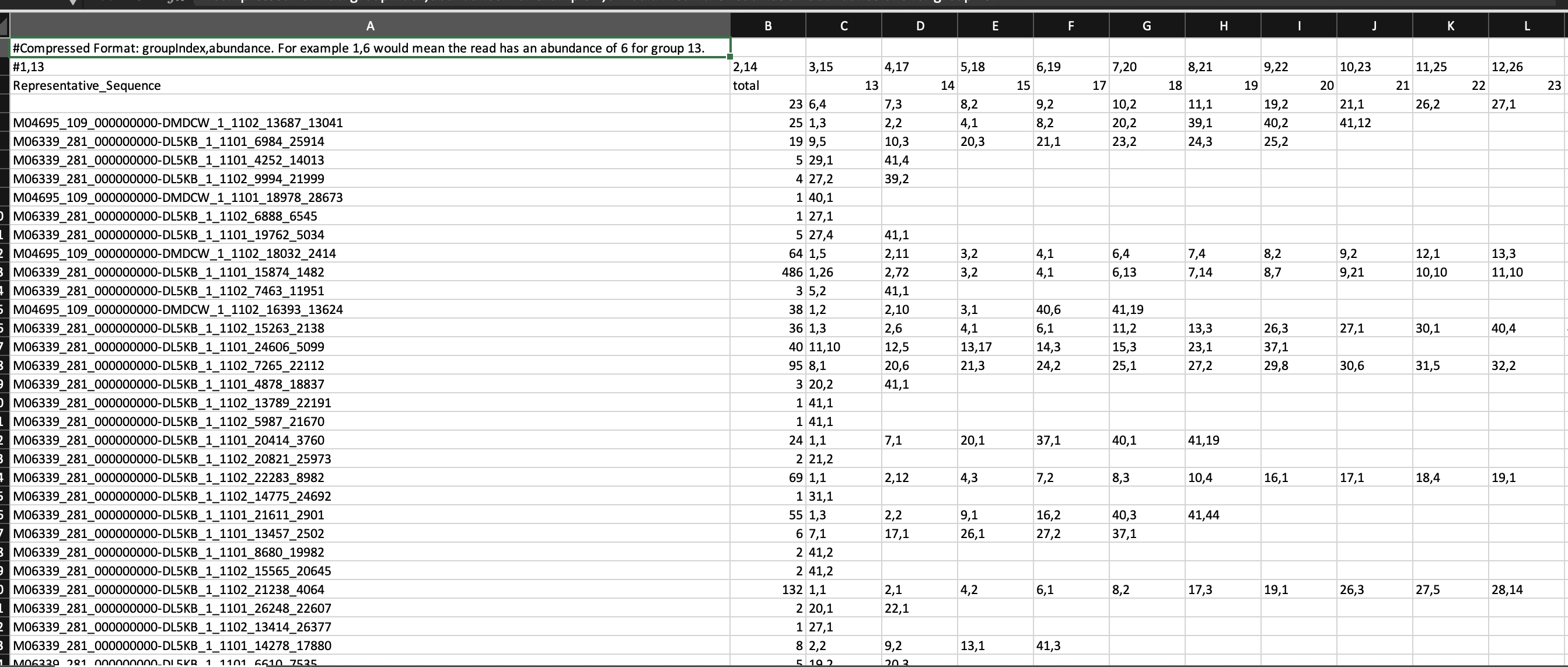
final.tx.1.rep.count_table.xls : output of the command to get the name of the sequence and search for it in the fasta file
final.tx.1.cons.taxonomy : phylotype analysis to match the .count_Table output with the taxonomy order
stability.trim.contigs.good.fasta : to look for the name and get the sequence.
If I doing the phylotype-based analysis, which count_table and fasta files would I use?
The screenshot you posted isn’t a mothur output file. The sequence fasta file is not likely in the same order as the oturep.fasta file. If you look at the MiSeq SOP you’ll see how to do phylotypes and get classifications for each phylotypes.
Pat
I apologize for the confusion, the screenshot was a combination of the outputs and how I tried to look for the species.
Attached is the command output.
Yes, I was able to classify for each phylotype and I was just wondering if there’s a way to know which phylotype corresponds to the sequence in the fasta file and know its sequence.
Thank you!
Thanks - can you post some of your output from get.oturep?
Pat
Yes, I tried the command with different files:
-
get.oturep(list=final.opti_mcc.list, count=stability.contigs.good.count_table, method=abundance)
Output File Names:
/Users/…/final.opti_mcc.0.03.rep.count_table
-
get.oturep(list=final.tx.list, count=final.count_table, method=abundance)
Output File Names:
/Users/…/final.tx.1.rep.count_table
Can you give it the corresponding fasta file and then share the outputted fasta file?
Pat
I tried both with the closest fasta file generated:
-
get.oturep(list=final.opti_mcc.list, count=stability.contigs.good.count_table, fasta=stability.trim.contigs.good.fasta, method=abundance)
Output File Names:
/Users/…/final.opti_mcc.0.03.rep.count_table
/Users/…/final.opti_mcc.0.03.rep.fasta
-
get.oturep(list=final.tx.list, count=final.count_table, fasta=final.fasta, method=abundance)
Output File Names:
/Users/…/final.tx.1.rep.count_table
/Users/…/final.tx.1.rep.fasta
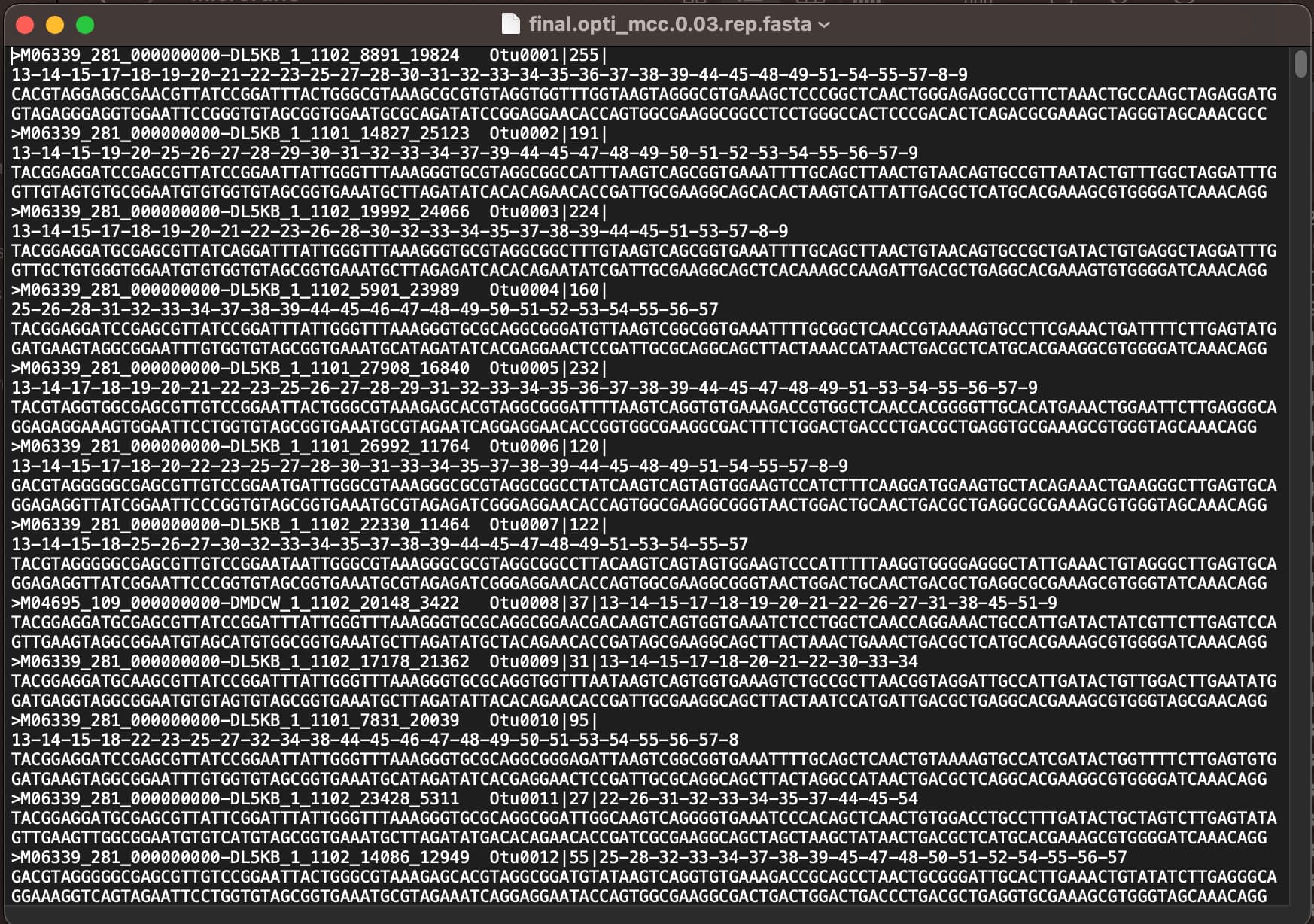
Based on this, the fasta file generated from the first command applied has the name of the sequence, the corresponding OTU, an abundance number (?), the samples it is found in, and the sequence.
I would use the first option since the fasta file has the complete sequence and use the OTU-based analysis outputs (shared and taxonomy files), correct?
I tried a couple of sequences that refer to the same OTU or Phylo using their respective shared files and I got same results for both so far.
That’s awesome, thank you for your help!
1 Like