Hi all,
I’m having a difficult time trying to figure out what has gone wrong with my filter.seqs. I get filtered alignment of 30.
Here is my code:
make.file(inputdir=., type=fastq, prefix=suis)
make.contigs(file=/scratch/shill09/files/suis.files,processors=32)
summary.seqs(fasta=suis.trim.contigs.fasta)
screen.seqs(fasta=suis.trim.contigs.fasta, group=suis.contigs.groups, maxambig=0, maxlength=500)
unique.seqs(fasta=suis.trim.contigs.good.fasta)
count.seqs(name=suis.trim.contigs.good.names, group=suis.contigs.good.groups)
summary.seqs(count=suis.trim.contigs.good.count_table)
pcr.seqs(fasta=silva.bacteria2.fasta, start=6388, end=25316, keepdots=F, processors=8)
rename.file(input=silva.bacteria2.pcr.fasta, new=silva.v34.prim2.fasta)
summary.seqs(fasta=silva.v34.prim2.fasta)
align.seqs(fasta=suis.trim.contigs.good.unique.fasta, reference=silva.v34.prim2.fasta)
summary.seqs(fasta=suis.trim.contigs.good.unique.align, count=suis.trim.contigs.good.count_table)
screen.seqs(fasta=suis.trim.contigs.good.unique.align, count=suis.trim.contigs.good.count_table, summary=suis.trim.contigs.good.unique.summary, start=17571, end=18912, maxhomop=8)
summary.seqs(fasta=suis.trim.contigs.good.unique.good.align, count=suis.trim.contigs.good.good.count_table)
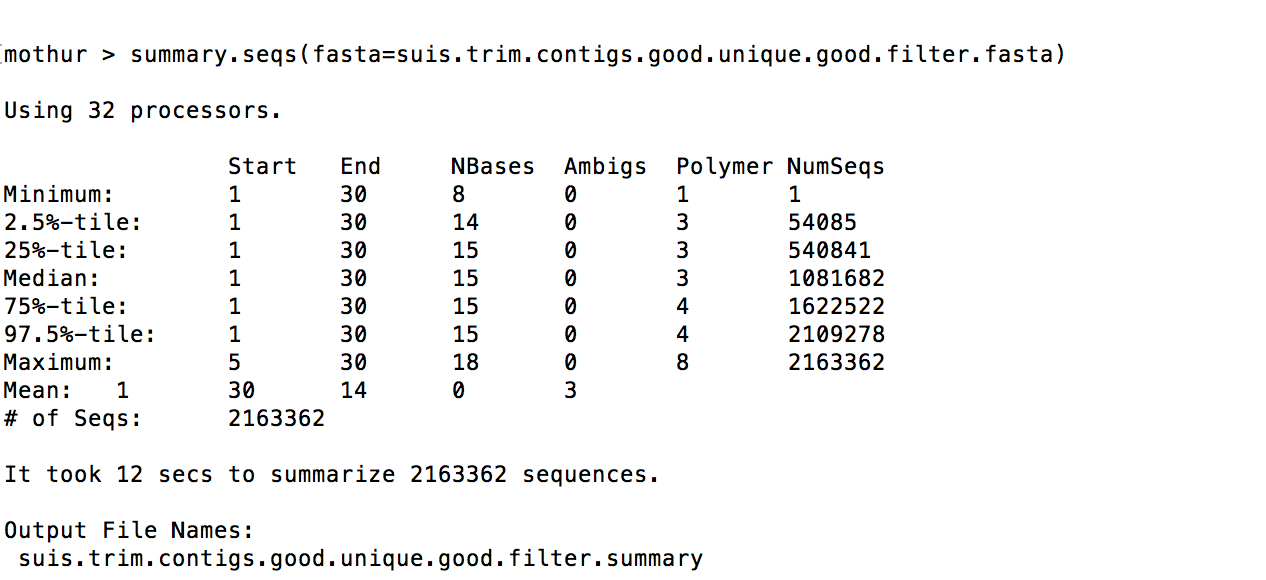
filter.seqs(fasta=suis.trim.contigs.good.unique.good.align, vertical=T, trump=.)
I’ve also attached the summary.seqs of the pcr.seqs, align.seqs, screen.seqs and filter.seqs!